
|
|
||
Our previous work gave us considerable experience in finding both previously known, and new motifs, some of which we subsequently verified experimentally. What follows are some pointers as to how to distil true motifs from noise.
It is first useful to study the nature of previously determined linear motifs. The majority of these are between 4-8 residues in length, have 2-4 specified (i.e. non-‘x’) positions, of which 1-3 are a single invariant amino acid. This can be seen in the figure below:
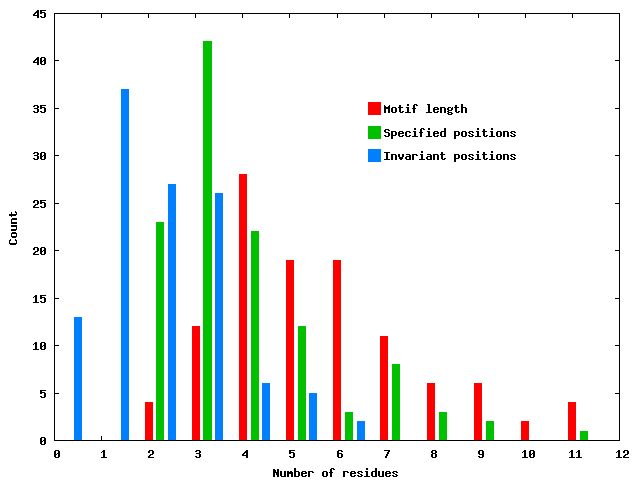
Known linear motifs also have different preferences for particular amino acids relative to globular proteins. These preferences also vary across different types of motifs (ligands, LIG; targeting signals, TRG; or post-translational modifications, MOD).
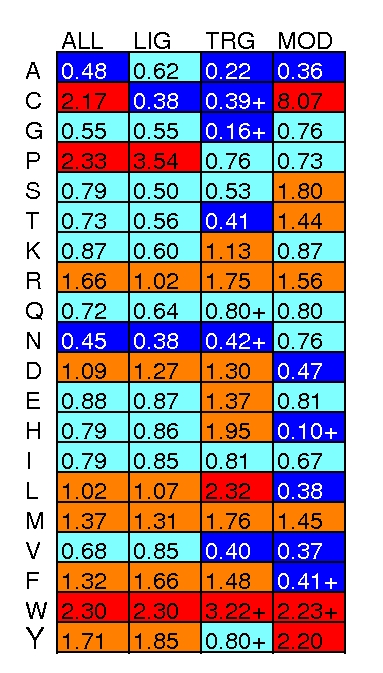
Proline features in many ligands, which is not surprising as many of the best known motifs contain it (e.g. SH3→PxxP and WW→PPxY). As would be expected, post-translational modification motifs contain more Cysteine, Serine, Threonine and Tyrosine residues than average sequences. Aromatic residues are also popular in all classes of motif. There are some curious differences between chemically similar amino acids. For example, Leucine and Methionine are common, but the β-branched aliphatic residues Valine and Isoleucine are not; Arginine is favoured, but not Lysine.
The range of P- and SCONS corresponding to real motifs is dependent on the particular species being considered. In our previous study we calculated maximum SCONS confidence values (p smaller than 0.001) for Yeast, Fly, Nematode and Worm (see here). These are reasonable starting points for motif hunting when looking in these species or their relatives, but are only a rough guide in practice, as real motifs can still occur with higher (less significant) values.
In practice, one should be prepared for the situation that a true motif might not be ranked first according to our metrics. This was the case even in our benchmark using sets of experimentally validated motifs. It tends to happen when motifs are very simple in nature, containing few residues, or those that are naturally abundant.
For more tentative motifs, we found it also useful to consider the number of times a motif is observed relative to the size of the set (beside the P- and SCONS values). In our hands, spurious motifs scored better when they occurred in a few members of a large set of proteins (e.g. 4 out of 30; most sets considered were 10 or fewer in size). This could be because large sets (e.g. 20 or more sequences) contain more putative false positives, and might also begin to deviate from the binomial distribution.
It is also always important to question the integrity of the set itself – that is do all the proteins really belong together, or are some more weak members than others? This is particularly so when one is dealing with interaction data from high-throughput techniques like the two-hybrid system. Having wrong sequences in a set does not mean that the approach will fail, but borderline motifs (in terms of significance), will end up scoring worse than they would in a cleaner set, and thus might be missed if the thresholds are set too high. Generally speaking, the cleaner the sequence set, the better the performance. If you have doubts about whether or not sequences belong in a set, try running the motif again after taking them out.
When referring to results from this server, please cite:
V. Neduva, R. Linding, I. Su-Angrand, A. Stark, F. de Massi, T.J. Gibson, J. Lewis, L. Serrano, R.B. Russell, Systematic discovery
of peptides mediating protein interaction networks PLoS Biology, 3, e405 2005.
PDF PubMed Full Text
We also studied the properties of known linear motifs in this paper:
V. Neduva, R.B. Russell, Linear motifs: evolutionary interaction switches, FEBS Lett., 579, 3342-3345, 2005.
PubMed
DILIMOT was developed by Victor Neduva and Rob Russell at EMBL, Heidelberg.
Please address questions, comments or bug reports toVictor Neduva